Тхе Бардет-Биедл синдром, такође Лауренце-Моон-Биедл-Бардет синдром (ЛМББС), је болест из поља цилиопатије која се јавља искључиво због наследности. Синдром се манифестује у облику вишеструких малформација које су потакнуте променама (мутацијама) на различитим локацијама гена или хромозомима.
Шта је Бардет-Биедл синдром?

© Цреатива Имагес - стоцк.адобе.цом
Клиничка слика коју су одредили лекари Моон и Лауренце, а касније Бардет и Биедл је болест у којој се дистрофија мрежнице јавља као медицински значајна карактеристика у комбинацији са другим симптомима. Због ове компликоване медицинске почетне ситуације тешко је одредити ББС болест. Ова клиничка слика први пут је медицински забележена 1866. године.
Четири прегледане особе имале су ретинитис пигментозу (ретинолошка дистрофија, РП) у комбинацији са параплегијом (спастичном парализом), као и хипогенитализмом (неразвијени генитални органи) и менталним хендикепом. 1920. године француски лекар Бардет описао је болест која се састојала од РП (дистрофија мрежнице), хипогенитализма, полидактилије и гојазности.
Прашки патолог Биедл такође је пронашао способност (менталну конфузију). Године 1925. истраживачи Веисс и Солис-Цохен сумирали су познате случајеве и описали клиничку слику Лауренце-Моон-Биедл-Бардет синдром.
узрока
У годинама које су уследиле, медицинска литература је све више указивала на то да су случајеви које су забележили Лауренце и Моон ретки посебан облик који се јавља само у појединачним случајевима заједно са ББС-ом. Последњи резултати медицинских истраживања сврставају Бардет-Биедл синдром у подручје цилиопатије (цилијарних болести).
Ове болести показују уобичајену неисправност такозваних цилија (мали процеси, антене), који се јављају на већини ћелија људског организма. За цилиопатије су карактеристични текући прелази и преклапања између различитих цилијарних болести.
Симптоми, тегобе и знакови
Главна карактеристика наследне дистрофије мрежнице је генерички израз који описује почетак губитка функције и накнадну дегенерацију (уништавање) фоторецептора. Доводе до прогресивног (прогресивног) губитка визуелних функција. Брзо прогресивни поремећаји вида обично се појаве врло рано код деце када су стари између четири и десет година. Осјећају се на различите начине, овисно о фоторецепторима на које утјечу.
Као "облик конусног штапића" са карактеристичним током ретинитис пигментоса (РП), болест потиче из периферне мреже ретина (спољна мрежница) и развија се у дегенерацију макуле (уништавање оштрог вида) прогресивним губитком видног поља.
Са гојазношћу (гојазност), тело показује патолошко нагомилавање масног ткива. У случају ББС-а, ненормално повећане накупине масти на ногама, стомаку, стражњици, рукама, грудима и куковима углавном се јављају као претилост дебла, при чему су дебљина, ноге и бедра посебно лоше погођени. Полидактилија је уочљив симптом и значајна карактеристика Бардет-Биедл синдрома. Откриће није лако јер се рудиментарна полидактија хируршки исправља након рођења.
Кс-зраке су у стању да дају даље информације. Полидактилија се може појавити са различитим знаковима, на пример као рудиментарни ножни прст или додатак прста. Прст или прст могу се формирати додатно или само делимично. Једнострана хексадактилија на стопалу и / или руци има додатну везу, билатерална хексадактилија се јавља и на ногама и / или на рукама.
Прсти или прсти који су израсли заједно (синдактилија) и скраћивање једног или више ножних прстију или прстију (брахидектилија) такође су знакови ББС-а. Само неколико пацијената имају сва четири захваћена екстремитета. Кашњење у менталном развоју је другачије. Само мали број оболелих показује озбиљну менталну ретардацију. Могућа је нормално обучена интелигенција.
Деца касно уче и говоре и ходају, а понекад показују и проблеме у понашању, попут анксиозних поремећаја. Компулзивна или аутистична понашања, низак праг фрустрације и нестабилна емотивност остале су могуће нуспојаве. Преферирају се познате, али промене се одбијају. Честе су неправилности у унутрашњим и спољним гениталним органима.
Остале промене су хипоспадија (отвор уретре изнад или испод, уместо на предњем делу пениса), трбушни или ингвинални тестиси, сужења уретре, сужење препуција и задња уретрална залистака. Код женских пацијената познати су вагинална атрезија (вагина није отворена), недостају отвори уретре и смањене унутрашње усне зубне усне.
Није неуобичајено да жене које су погођене имају нередовите менструалне циклусе. Промене бубрега су честе нуспојаве. Налаз зависи од прегледа доњег мокраћног тракта и бубрега помоћу ултразвука (сонографија).
Дијагноза и ток болести
Бардет-Биедл синдром (ББС) има шест главних симптома, али не појављују се заједно у сваком случају. Лекари претпостављају одговарајући налаз ако су присутна најмање четири главна симптома. Алтернативно, постоји велика вероватноћа да је болест присутна ако пацијент има три главна симптома и два секундарна симптома.
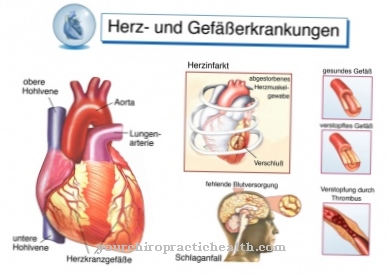
Шест главних симптома су дистрофија мрежнице, гојазност (абнормално накупљање масног ткива, прекомерна тежина), полидактилија (вишак ножних прстију и / или прстију), ментална заосталост (застој менталног развоја), хипогенитализам (неразвијени генитални органи) и бубрежна болест. Нискофреквентни секундарни симптоми укључују застој говора, говорни дефицит, срчане малформације, атаксију (поремећена координација покрета), астму, дијабетес мелитус (дијабетес), Црохнову болест (упала великог и / или танког црева), дисплазију ребара и краљежака и кифосколиозу (вертебрална анемија) на.
Компликације
Са синдромом Лауренце-Моон-Биедл-Бардет, погођени обично пате од губитка видне функције. Губитак не настаје нагло, већ постепено. У најгорем случају погођени ће остати потпуно слепи, што се обично више не може лечити.
Посебно код младих и деце слепило може довести до озбиљних психолошких тегоба или чак депресије. Пацијенти су у свакодневном животу јасно ограничени и пате од знатно смањеног видног поља. У многим случајевима синдром Лауренце-Моон-Биедл-Бардет такође доводи до проблема у понашању, тако да деца посебно могу патити од силовања или задиркивања.
Синдром такође значајно касни и ограничава развој деце. Могу се јавити и анксиозни поремећаји. Није реткост да синдром Лауренце-Моон-Биедл-Бардет доведе до психолошких тегоба и депресије код родбине или родитеља. Узрочно лечење синдрома Лауренце-Моон-Биедл-Бардет нажалост није могуће.
Неке жалбе могу бити ограничене. Потпуно позитиван ток болести ипак није уследио. Синдром не смањује животни век пацијента. У неким случајевима погођени понекад требају помоћ других људи у свакодневном животу.
Када треба ићи код лекара?
Пошто је синдром Лауренце-Моон-Биедл-Бардет наследна болест, дијагноза се може поставити у матерници. Најкасније након рођења, потребно је консултовати лекара ако се примете типични симптоми, попут поремећаја вида или гојазности. Малформације ножних прстију и прстију такође су јасан показатељ болести.Родитељи који примете симптоме код свог детета треба одмах да обавесте педијатра.
Опсежан преглед пружа информације о болести. Након тога, терапија се обично започиње директно, а састоји се од различитих третмана ортопеда, неуролога, офталмолога, интерниста и терапеута, као и физиотерапеута. Даљње посете лекару су неопходне ако лечење нема жељени ефекат. Савет лекара је потребан и у хитним ситуацијама, на пример ако дете падне као последица малформације или изненада има напад. Ако болесна особа покаже знакове емоционалне нелагоде, родитељи се морају консултовати са одговарајућим терапеутом. Старија деца могу да контактирају школског психолога заједно са родитељима и разговарају о одговарајућим мерама.
Терапија и лечење
Ова болест се јавља на основу аутосомно рецесивног насљеђивања, што значи да обје копије (алела) ББС гена показују промјену (мутацију). Пацијентови родитељи су „мешовити“ и сваки носи модификовани и непромењени алел одговарајућег гена. Они немају болест. Деца се разболе само ако им отац и мајка пренесу мутирани алел. Уз додатну децу вероватноћа понављања је 25 процената.
Опција каузалне терапије још није позната, јер се одређени симптоми болести још увек не могу у потпуности доделити различитим генетским променама. Симптоми и њихове манифестације појављују се различито чак и код болесне сестре. Пошто је карактеристична пуна слика ББС-а присутна само у ретким случајевима, посебно код мале деце, одговарајућа дијагноза је тешка.
Због често присутних олигосимптоматских симптома, код којих се јавља врло мало атипичних и тек благо изражених симптома, друге могуће клиничке слике морају се узети у обзир у диференцијалној дијагнози. Промене у истом гену могу довести до различитих клиничких слика, на пример Јоуберт, Бардет-Биедл или Мецкел-Грубер синдром.
Изгледи и прогноза
Прогноза за присуство синдрома Лауренце-Моон-Биедл-Бардет је углавном лоша, јер су вишеструке малформације урођене и неизлечиве. Ако се појаве четири од шест главних симптома, дијагноза Лауренце-Моон-Биедл-Бардет синдрома је потврђена. Бројни секундарни симптоми додају главним симптомима. Ово укључује пузеће слепило.
Због сложености симптома нема изгледа за излечење. Постоји само осредња шанса за приметно олакшање симптома. Број могућих малформација и поремећаја у синдрому Лауренце-Моон-Биедл-Бардет је толико велик да је наследну болест тешко лечити. У сваком случају, на ток генетске болести не може се утицати. Међутим, садашњи симптоми се могу делимично ублажити.
Међутим, лоша укупна прогноза не смањује очекивани животни век погођених људи. У старијем добу и након слепљења, погођени могу трајно да зависе од помоћи или неге. Кроз интердисциплинарне медицинске напоре, многи оболели од синдрома Лауренце-Моон-Биедл-Бардет могу доживети нешто блажи ток болести.
Растући видни проблеми представљају тешко лечење и проблематичан део болести, а све већа оштећења вида већ се јављају код погођене мале деце. Они се временом погоршавају. Проблеми са видом не морају довести до слепила код свих који су погођени. Психолошке посљедице синдрома Лауренце-Моон-Биедл-Бардет обично се могу добро лијечити.
превенција
Превенција у смислу спречавања ове болести није могућа. Важно је редовно праћење симптома и пратећих симптома. Могући терапеутски приступи су поновљени прегледи крвног притиска и рада бубрега, савети о исхрани, физиотерапија и радна терапија, као и логопедска терапија.
Послије његе
У већини случајева, они који су погођени синдромом Лауренце-Моон-Биедл-Бардет немају посебне могућности праћења, тако да се о овој болести мора обратити лекару и консултовати га врло рано. По правилу не може доћи до само-излечења, па је лечење лекара увек неопходно.
Пошто је синдром Лауренце-Моон-Биедл-Бардет наследна болест, дотична особа треба да има спроведен генетски преглед и савет уколико жели да има децу, тако да синдром Лауренце-Моон-Биедл-Бардет не пређе на њихово потомство се преноси даље. У многим случајевима, погођени зависе од хируршких интервенција за ублажавање малформација и деформитета.
Овде особа која је погођена дефинитивно би се требало одмарати након захвата и водити рачуна о свом телу. Треба избегавати вежбање или друге физичке и стресне активности у сваком случају како се не би непотребно оптерећивало тело. Пошто синдром Лауренце-Моон-Биедл-Бардет такође може довести до ненормалног понашања, родитељи би требали подржати и охрабрити дете у развоју. Љубавни и интензивни разговори са дететом су такође неопходни како би се спречили психички поремећаји или депресија.
То можете и сами
Лауренце-Моон-Биедл-Бардет синдром има различите симптоме, при чему пацијент често пати од оштећене видне функције. Чак и код деце, уобичајена способност вида почиње да се погоршава, тако да су родитељи ти који дијете представљају лекару и на тај начин убрзали дијагнозу. На овај начин се болест може брзо лечити, мада су до сада могућности лечења биле само симптоматске природе.
Поремећаји вида све се више повећавају код болесне деце и на тај начин значајно нарушавају свакодневни живот, тако да се квалитета живота оболелих смањује. Зато што проблеми са видом развијају бројне потешкоће за пацијента када похађа школу, у слободно време и физички интегритет. Ризик од несрећа такође се значајно повећава, на пример у друмском саобраћају. Због тога родитељи прате своју болесну децу кад год је то могуће или ангажују неговатељско особље како пацијент не би остао да се брине за себе.
У неким случајевима се болест шири до тачке слепила. Пошто је такав развој догађаја већ очигледан унапред, пацијенти се припремају за њега. Родитељи редизајнирају животни простор тако да у њему не постоје извори опасности за слабовидну особу. Поред тога, слепе жртве уче како користити дугачак штап како би се самостално кретале изван властитог дома.










.jpg)




.jpg)

.jpg)