Укупно је 45 различитих болести лизосомалног складиштења, које су хетерогена група урођених метаболичких болести. Људи који пате од било које од ових болести имају генетску ману. Све болести складиштења имају једно заједничко: одређени ензим је или одсутан или је само дјелимично функционалан.
Шта је болест лизосомалног складиштења?

© десигнуа - стоцк.адобе.цом
Ове болести конгениталног складиштења су ретке, јер је погођено мање од пет на 10.000 људи. Разне болести имају врло различит ток, а симптоми могу веома варирати.
Најпознатији облици болест лизосомалног складиштења су Фабријева болест, Гауцхерова болест, Помпеова болест и мукополисахаридоза (МПС). Често их називају „сирочетима медицине“, јер пут до конкретне дијагнозе и одговарајуће терапије може бити јако дуг. Понекад могу проћи године док они који су погођени могу сазнати шта се са њима дешава.
узрока
За болести складиштења лизосома карактеристични су одређени облици наследних метаболичких болести. Пацијенту недостаје важан ензим који обезбеђује несметану метаболичку равнотежу. У мање израженом облику, овај ензим барем није присутан у довољним количинама.
Задатак ензима је да одлажу загађиваче и отпадне материје које се у организму човека накупљају путем метаболизма преко лизосома, или да их поново обрађују на начин да се симптоми не појаве.
Ако постоји недостатак ензима, овај несметано циклус одлагања више није загарантован. Штетне материје се слежу у ћелијама и нарушавају метаболички циклус. У почетној фази поремећаји немају приметни ефекат, постоји само неколико ограничења. Међутим, ако овај метаболички поремећај остане не лечен као резултат недостатка ензима, симптоми се умножавају јер ћелије постају веома проширене.
Симптоми, тегобе и знакови
У најгорем случају ови пропадају. Последице су оштећења костију, нервног система, слезине, бубрега, мишића или срца. Због смањене или одсутне ензимске активности, Фабријева болест изазива складиштење масти (глоботриаосилцерамид, Гб3) у ћелијама. Ове нежељене наслаге могу довести до јаких болова у ножним или прстима, можданог удара и оштећења бубрега.
Дијагноза и ток болести
Ова клиничка слика истовремено утиче на различите системе: крвне судове, бубреге, срце и нервни систем. Аутосомно рецесивна Гауцхерова болест изазива мутацију ензима „бета-глукоцеребросидаза“ и доводи до накупљања супстрата унутар ћелија, посебно у макрофазима (фагоцитима) који припадају ретикуло-ендотелном систему. Крвна слика се мења, јетра и слезина су повећане, а кости боле.
Болест је прогресивна и углавном је етничка, јер се јавља у већини случајева код људи јеврејског порекла. Помпеова болест је позната и као "недостатак киселе малтазе". Клиничка слика припада групи гликогенезе типа ИИ. Погођеним особама недостаје ензим "алфа-1,4-глукозидаза" (кисела малтаза) или није доступан у довољним количинама. Због поремећеног распада гликогена у мишићима, пацијенти пате од уништавања мишићних ћелија у виду складиштења шећера.
Мукополисахаридоза типа И (МПС), такође позната и као Хунтерова болест, има различите клиничке узроке. Хурлерова болест је најтежи облик, а Сцхеиеова болест је на крају клиничке патогенезе. Постоје прелази различитих карактеристика између ова два облика прогресије. Најупечатљивије својство је оштећење разградње угљених хидрата који се накупљају у лизосомима ћелија.
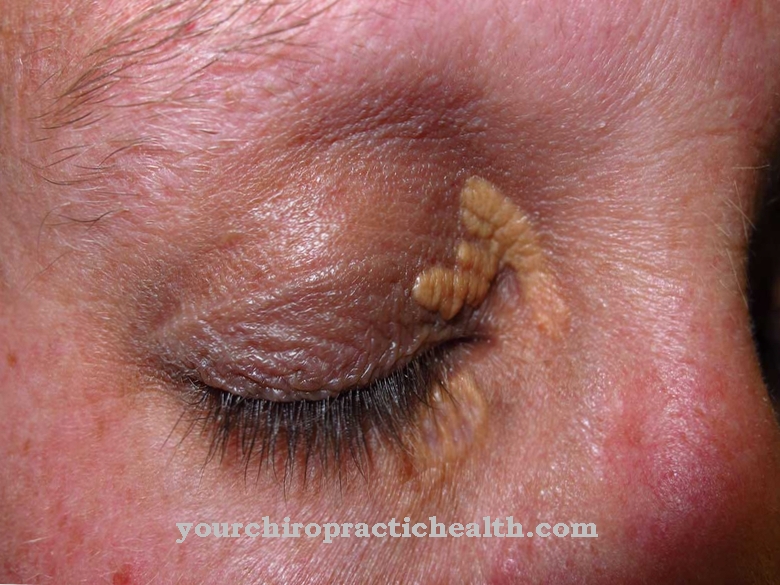
Болесници са ловачком болешћу могу да имају кратак раст, увећану слезину и јетру, грубе карактеристике, задебљану кожу, проширени језик и отежано дисање. Уз то, скелет се често мења у подручју карлице, кичме, костију руку и лобање. Могуће су пупчане и [[ингвиналне киле].
Компликације
У већини случајева симптоми или компликације код ове болести се јављају врло касно. Због тога се дијагностикује касно, што онемогућава рано лечење у већини случајева. Без лечења, разне болести и оштећења унутрашњих органа настају како болест напредује.
Поготово су погођени бубрези, јетра и слезина. На срце може бити погођена и ова болест, која у најгорем случају може довести до срчане смрти. Надаље, долази до оштећења бубрега, а обољели често пате од болова у ножним или прстима. Парализа може да се деси и ако је мозак оштећен овом болешћу. Јетра и слезина се могу повећати и проузроковати јаке болове.
Није неуобичајено да кости оболеле особе буду ломљиве и такође болне. Лечење ове болести показује се тешким. У многим случајевима се животни век особе која је погођена значајно смањује. Обично нема посебних компликација приликом употребе лекова. Међутим, позитиван ток болести не може се гарантовати у сваком случају.
Овде можете пронаћи лекове
➔ Лекови против боловаКада треба ићи код лекара?
Губитак косе, проблеми са зглобовима и поремећаји органа могући су знакови болести лизосомалног складиштења. Посета лекару се препоручује ако се симптоми понављају или се појављују изненада, без проналаска узрока. Ако су симптоми повезани са претходно дијагностицираном оштећењем ензима или другом озбиљнијом болешћу, потребно је консултовати одговорног лекара. Нелијечена болест складиштења може довести до деменције, неплодности, неуропатија и других компликација, од којих су неке опасне по живот. Стога би требало испитати све могуће симптоме, чак и ако нема одређене сумње.
Симптоми болести лизосомалног складиштења могу се појавити у фазама или се развијају подмукло, али увек захтевају преглед и лечење. Погођене људе је најбоље разговарати директно са својим породичним лекаром или интернистом. Стварна терапија се обично одвија у специјалистичкој клиници за унутрашња обољења, мада се физиотерапија или психотерапија могу повезати у зависности од симптома. Нарочито су терапијске мере индициране због често негативног тока болести.
Терапија и лечење
У зависности од тога како се рано постави адекватна дијагноза, ове насљедне болести могу се врло добро лечити надомјесном ензимом, тако да особе које пате имају далеко мање симптома и самим тим бољи квалитет живота. Ова замена терапија се користи у складу са клиничком сликом.
Људима који пате од Гауцхерове болести недостаје „ензим ß-глукоцеребросидаза“, који се производи биотехнолошки и улива у тело пацијента. Лизосоми делују ефикасно и способни су да апсорбују материје из непосредног окружења. Из тог разлога, вештачки коришћени ензими су модификовани на такав начин да се могу идеално доставити лизосомима.
Макрофаги (фагоцити) разграђују глукоцереброзиде који су се акумулирали у ћелијама. Ова терапија се може упоредити са инзулинском терапијом за дијабетес мелитус, с разликом што то није хормон који недостаје, већ ензим који се не испоручује. Тело редовно разграђује све супстанце, укључујући испоручени вештачки ензим.
Због овог редовитог разлагања супстанце, пацијенти морају да редовно подносе ову инфузијску терапију до краја свог живота. Ензим замјенска терапија не дјелује симптоматски, него се директно бори против узрока насљедне болести. Лекари ову терапију називају узроком. Принципи терапије се користе за све четири горе поменуте болести складиштења.
Помпе пацијенти се такође лече инфузијском терапијом. Код ове болести, непостојећи ензим "киселина алфа глукозидаза" се обезбеђује и помаже у разградњи гликогена који се накупља у лизосомима мишића. У болесника са болешћу типа „мукополисахаридоза тип И“, лизосомални ензим „алфа-идуронидаза“ није присутан или није присутан у довољним количинама. То је једно од најређих болести складиштења у коме се молекули шећера накупљају у органима и ткивима.
Ако је процес нормалан, ензим разграђује мукополисахариде. Молекули шећера су дуго везани и учествују у развоју потпорног и везивног ткива, на пример кости, коже, зглобова и хрскавице. Ако је нормалан ток разградње поремећен због недостатка ензима, у појединим ћелијама акумулирају се патолошки гликозаминогликани (ГАГ). Будуће могућности терапије усмерене су на узимање таблета.
Изгледи и прогноза
Прогноза за болест складиштења је лоша. Нађено је да је генетска диспозиција узрок здравственог поремећаја. Законски захтеви забрањују лекарима и научницима да мењају људску генетику. Из тог разлога, болест остаје доживотно и нема изгледа за опоравак.
Лекар који се бави се концентрише на лечење симптома који се јављају. Ако се не лечи, с временом ће се повећавати разне жалбе. Коштани систем је оштећен и настају проблеми органа. У најгорем случају, унутрашњи органи ће неправилно функционисати и на крају њихова функција неће успети. Ово прети дотичној особи прераном смрћу.
Изазов болести лежи у дијагнози. Код великог броја пацијената уочљиве и снажно уочљиве притужбе јављају се тек касније у животу. Као резултат тога, генетски поремећај остаје дуго непримећен, а рано лечење болести је тешко. Што се постави дијагноза, то је неповољнији даљи ток. У поодмаклој фази болести унутрашњи органи или зглобови су већ озбиљно оштећени. Потребне су хируршке интервенције и ако болест лоше напредује, само један орган донора може спасити живот оболелој особи. Стога је рано лечење од суштинског значаја за побољшану прогнозу.
превенција
Пошто је реч о урођеној генетској оштећењу која спречава експресију ензима, ова болест се не може лечити превентивно. Међутим, најновија достигнућа у генетском инжењерингу могла би да пруже приступ у овој области.
Послије његе
Уз ову болест, људи пате од низа различитих компликација и болести. У правилу, сви они имају врло негативан утицај на квалитет живота особе на коју погађају, тако да би требало дијагнозу поставити врло рано.Што се раније консултује са лекаром, то је бољи даљи ток ове болести обично.
Тежина ове болести може јако варирати, тако да опште предвиђање често није могуће. Они који пате трпе тешко оштећење унутрашњих органа. Превасходно су погођени бубрези и срце, тако да дете може умрети у првих неколико дана ако се симптоми не отклоне на време. Такође постоје депозити масти у различитим деловима тела.
Посебно су погођени прсти и ножни прсти, што може довести до значајно смањене естетике код особе на коју погађају. У правилу, оштећење бубрега и мозга настаје у даљем току, тако да особа која је погођена умре као резултат ове штете. Родитељи и рођаци такође често пате од депресије или других менталних поремећаја због болести.
То можете и сами
Болести лизосомалног складиштења често захтевају интензивну медицинску негу. Често нема довољно могућности за самопомоћ. Родитељи погођене деце често доживљавају снажан стрес у свом кућном окружењу, јер им је дете потребно стална нега и пажња.
Клиничке слике појединачних болести складиштења су различите. Постоје и лаки и веома тешки облици. Један пример је Гауцхерова болест. Помоћ родитеља често је ограничена на храњење детета тешко инвалидног стања. У блажим случајевима животни век може бити готово нормалан. Ипак, стални медицински надзор је потребан да би се избегли могући компликације. Редовна физичка активност једна је од пратећих терапија која се може изводити и код куће. Надаље, мора се уговорити темељита провера прегледа рака. Ово захтева сталне посете лекару са својим дететом од родитеља. Исто се односи и на остале лизосомске болести складиштења.
У случају неких болести, поред физичких оштећења, могу се јавити и ментална оштећења, која и даље захтевају посебну подршку. Код блажих облика одређених болести, попут Хунтерове болести, у почетку се јављају само скелетне промене и дисморфизам лица. Међутим, овде је погођени пацијент у стању да води самосталан живот. Међутим, овде су потребни и стални лекарски прегледи како би се искључили могући компликације попут затајења срца или респираторних болести. Пацијент се може суочити са психолошким стресом изазваним физичким деформацијама путем психолошког саветовања.

.jpg)
.jpg)

.jpg)


.jpg)







.jpg)