На Фрасер синдром то је веома ретка генетска болест коју карактерише неколико физичких малформација. Обим појединих малформација није уједначен, тако да поред случајева мртворођене и деце која умиру одмах након рођења, постоје и они који су погођени нормалним животним веком. Терапија зависи од врсте малформације.
Шта је Фрасер синдром?

© владимирцарибб - стоцк.адобе.цом
Фразеров синдром први је пут описао 1962. године британски хумани генетичар Георге Р. Фрасер. Због једног од главних симптома, затворене пукотине очних капака, ова болест се у почетку звала криптофталмос синдром. У наредним годинама је дијагностицирано више случајева.
У веома многим случајевима, поред криптофталмоса, пронађене су и адхезије суседних прстију или ножних прстију (синдактилија). Због тога се често користио синонимни термин криптофталмос синдромтилија. Остали синоними болести укључују Меиер-Сцхвицкератхов синдром, Уллрицх-Феицхтигеров синдром или Фрасер-Францоисов синдром.
Израз Фрасер синдром, назван по америчком генетичару људске генерације Вицтор Алмон МцКусицк, постао је главни појам. Укупно постоји око 150 случајева случаја овог синдрома. Утврђено је да постоје различити облици појединачних малформација. Неки обољели само пате од криптофталмоса без даљњих физичких деформација.
У осталим случајевима јављају се разни дисморфизми. Болест има различите главне симптоме и нуспојаве, с тим да се животни век појединих пацијената такође разликује. 25 процената погођене деце је мртворођено. Још 20 процената пацијената умре од неправилности у раду бубрега или гркљана у првој години живота. Међутим, већина људи има нормалан животни век.
узрока
Аутосомно рецесивна мутација гена је могући узрок Фрасеровог синдрома. У већини случајева постоји генетска оштећења гена ФРАС1, који се налази на хромозому 4. Остали гени који су погођени су ГРИП1 на хромосому 12 или ФРЕМ2 на хромозому 13.
ФРАС1 ген кодира протеин који припада ванћелијском матриксу. Мањак одређених протеина у ванћелијском матриксу доводи до неравнотеже у процесима раста током ембриогенезе. Насљеђивање свих погођених гена је аутосомно рецесивно. То значи да болесна особа мора бити хомозиготна за два неисправна гена.
Ако је мутирани ген присутан само једном, болест неће избити. Синдром се може наследити само ако оба родитеља имају барем један неисправан ген. Стога се овај синдром накупља у потомству сродних родитеља који имају овај одређени генетски недостатак.
Симптоми, тегобе и знакови
Фразеров синдром карактерише појава различитих главних и мањих симптома. Потврђена дијагноза може се претпоставити ако се појаве или два главна симптома и један секундарни симптом или један главни симптом и четири секундарна симптома.
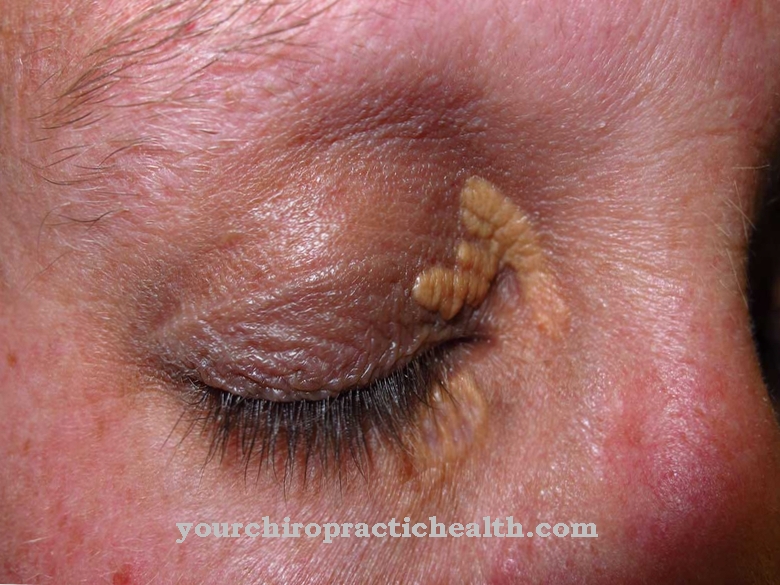
Главни симптоми су недостатак расцјепа очних капака (криптофталмос), неправилности органа урогениталног тракта као што су бубрези или генитални органи, спојеви суседних прстију или ножних прстију (синдактилија) и неправилни или нестали сузни канали.
Секундарни симптоми укључују слабост главе (микроцефалија), широко олакшање очију (хипертелоризам), сужење или одсуство гркљана, незрело плућно плуће (плућна хипоплазија), проширење симфизе стидне кости и скелетне абнормалности. Такође се примећују малформације ушћа, усана или непца, као и померање пупка или брадавице.
Когнитивно оштећење раније се претпостављало као још један симптом. Међутим, то није потврђено. Прогноза болести зависи од било каквих малформација бубрега, гркљана или плућа. То узрокује високу стопу мртворођених и смртност новорођенчади. У супротном, животни век није ограничен.
Дијагноза и курс
Поред анамнезе, дијагноза се углавном односи на комбинацију главних и секундарних симптома. Главни фокус је на испитивању очију, бубрега, гркљана и плућа. Фразеров синдром може се дијагностицирати пренатално уз помоћ ултразвучних прегледа. Могу се открити ехогена плућа која указују на сужавање или затварање гркљана.
Течност која се непрестано формира више се не може вратити у плућа због сужавања гркљана и тако формира загушење, што је такође познато и као фетални ЦХАОС (конгенитални синдром опструкције дишних путева). „Конгенитални синдром опструкције високих дисајних путева“ значи „сужавање горњих дисајних путева“.
Ово стање може довести до мртвородјености и сматра се медицинском индикацијом за могући прекид трудноће. Пренатална дијагноза такође укључује људске генетске прегледе помоћу узорковања хорионских ресица или амниоцентезе.
Компликације
Фразеров синдром заснован је на генетској оштећењу, па узрочно лечење није могуће. Због реткости синдрома и високе стопе смртности оболелих (скоро четвртина оболелих умре пре рођења, даљња четвртина у првој години живота), терапијски приступи и компликације нису адекватно описани.
Преживели се обично лече хируршки, зависно од тога како су погођени. Због тога су најчешће компликације у лечењу Фрасеровог синдрома генерални ризици повезани са операцијом. Укључују тромбозе вена на ногама (крвни угрушци у ногама, оклузија плућног суда (плућна емболија), повреде живаца и инфекције. Постоперативно, постоји повећан ризик од поремећаја зарастања рана, укључујући гангрену).
Ако се не лечи, уобичајени симптоми Фрезеровог синдрома могу озбиљно утицати на општи живот оболелих. Нелечена микрофталмија (или анофталмија) резултира потпуним губитком вида који може бити слаб. Ако се козметичка корекција очију не врши (нпр. Уметање очне протезе или корекција јаког шкљоцања), главне су последице психолошка оштећења до озбиљности депресије.
Ако синтактија (прсти и / или ножни прсти који су израсли заједно) остане необрађена, моторичке способности су озбиљно нарушене - зависно од озбиљности, хватања, држања оловке или ходања тешко је могуће. Бубрежна агенеза често повезана са Фрезеровим синдромом, ако се не лечи, доводи до смрти пацијента.
Када треба ићи код лекара?
Физичке неправилности које одступају од норме увек морају да разјаве лекар. Људско тело има основну структуру, чија девијација указује на болест. Лепљење, малформације или израслине сматрају се необичним и треба их испитати лекар. Ако су у људском скелетном систему промене, неопходна је посета лекара, чак и ако нема даљег притужбе.
Лекар мора проценити малформације, окоштавање или ограничења у опсегу кретања. Ако се ножни прсти и прсти не могу померати појединачно, ако су се променили због другачијег облика или ако погођена особа има руке и ноге у облику мреже, препоручљиво је да се обратите лекару.
Ако је интерпупиларна удаљеност необична, гркљан недостаје или се у грлу сужавају, потребан је лекар. Деформитете лица, посебно ушију, очију, носа или усана, треба прегледати медицински. Ако се непце промени, потребан је и лекар, јер то може довести до проблема са уносом хране.
Ако су пупак или брадавице у различитим положајима на телу, ове карактеристике треба представити лекару. Одступања у могућим покретима зглоба или визуелне промене зглобова сматрају се необичним и треба их прегледати лекар. Ако неки од појединачних система не раде због постојећих деформација, потребан је љекар.
Љекари и терапеути из ваше околине
Лечење и терапија
Малформације бубрега и гркљана главни су узроци смрти код Фреусеровог синдрома. Стога, ако дође до неправилности гркљана након рођења, одмах се мора извршити такозвана трахеотомија. Током трахеотомије креира се хируршки приступ душнику како би се осигурало прозрачивање пацијента.
У каснијим годинама, овисно о ситуацији у којој се налазите, обнављање функција може се ревидирати. Нажалост, одсуство оба бубрега није компатибилно са животом. У овом случају лечење више није могуће. Ипак, с једностраном бубрежном агенезом задржава се функција једног бубрега. Надаље, циљано је хируршко отварање фисура на капку. Међутим, ова операција има смисла само ако су очне јабучице присутне.
Изгледи и прогноза
Фразеров синдром је изузетно ретко дијагностикована болест широм света. Тешко да постоји више од 150 документованих случајева у којима је постављена дијагноза. Већина пацијената до сада има кратак животни век. Због озбиљних малформација, умиру у матерници, одмах по рођењу или недуго након рођења. Ова деца су патила од тешких малформација гркљана или бубрега.
Будући да се физичке малформације јављају појединачно, постоје и болесни људи који не примете значајно смањење животног века. Многе мутације се могу оптимизирати у хируршким интервенцијама након рођења, тако да је могуће побољшање квалитета живота.
Пацијент има добру прогнозу за неправилности пукотина очних капака, сузних канала или адхезија прстију и ножних прстију. Корективне мере могу се предузети већ у првих неколико година живота у оквиру процеса раста и развоја. Жељене промене врше се најкасније с крајем физичког раста.
Потпуно излечење није могуће код генетске болести. Људски ДНК се не може и не сме мењати. Упркос томе, симптоми се могу лечити добро и на адекватан начин помоћу доступних могућности терапије и лечења. Малформације гениталних органа или органа урогениталног тракта могу довести до секундарних болести. У неким случајевима пацијент је зависан од органа донора.
превенција
Фразеров синдром је генетски и често се јавља у сродним браковима. Ако постоји породична анамнеза, треба користити генетско саветовање и, ако је потребно, ДНК тест ако желите да имате децу. Поред тога, доступне су методе пренаталног прегледа које се могу користити за пренаталну дијагностику Фразеровог синдрома.
Послије његе
По правилу, не постоје посебне опције праћења за оне који су погођени Фрасеровим синдромом, пошто се ради о генетској болести. Пацијент је зато зависан од чисто симптоматског лечења ове болести у циљу спречавања даљих компликација и избегавања смањеног животног века. Не мора се очекивати само излечење код Фрасеровог синдрома.
Уз симптоматско лечење, обично се може задржати нормалан животни век. У већини случајева лечење ове жалбе се решава хируршким путем. Нема посебних компликација и вентилација се поново може обезбедити. Након овог напорног поступка, особа која је погођена мора се одмарати и бринути о телу.
Овде се не смеју обављати напорније или стресније активности. Строго се треба придржавати мировања. Будући да Фразеров синдром такође може оштетити бубреге, редовни прегледи од стране интерниста су врло корисни да се то оштећење спречи.
Очи такође треба редовно прегледавати и лечити. Будући да Фрасер синдром такође може негативно утицати на психу особе која је погођена, подршка и помоћ пријатеља и породице је врло корисна у спречавању компликација.
То можете и сами
Скраћени животни век, који је често повезан са Фрезеровим синдромом, представља посебан изазов за болесне и рођаке. Будући да је психолошки стрес изазван овом прогнозом веома висок, требало би да се нађе начин да се самостално нађе у свакодневном животу поред конвенционалне медицинске подршке како би се могли добро носити са теретом.
Отворена комуникација са члановима породице и пријатељима помаже у разјашњењу међусобних забринутости и страхова. У многим случајевима треба тражити терапијску подршку да не би дошло до даљих болести из психолошког притиска.
Размјене група самопомоћи или форума могу вам помоћи да добијете информације и савјете о суочавању са болешћу у свакодневном животу. О искуствима и искуствима разговара се са другим погођеним људима. То помаже у преради догађаја и јача психу.
Поред тога, процеси опуштања могу се користити за враћање унутрашње равнотеже. Уз Ки Гонг, аутогени тренинг, медитацију и јогу, стрес се може смањити и ојачати ментална снага. Појединачне малформације значе да је пацијент зависан од свакодневне помоћи. То резултира реструктуирањем које има утицаја на остале чланове породице. Ипак, треба одржавати позитиван став и проводити време заједно за пријатне активности.
.jpg)

.jpg)

.jpg)


.jpg)







.jpg)